Энергетический обмен в миокарде при сердечной недостаточности

Сердечная недостаточность – патологическое состояние, заключающееся в неспособности сердца обеспечить органам и тканям количество крови, соответствующее их метаболическим потребностям организма и необходимое для нормального функционирования организма. Сердечная недостаточность кровообращения развивается в результате ослабления сократительной функции миокарда. Причинами его являются:
1) переутомление миокарда, вызванное рабочей перегрузкой сердца (при пороках сердца, повышении периферического сопротивления сосудов – гипертонии большого и малого круга кровообращения, тиреотоксикозе, эмфиземе легких, физическом перенапряжении);
2) непосредственное поражение миокарда (инфекции, бактериальные и небактериальные интоксикации, недостаток субстратов метаболизма, энергетических ресурсов и пр.);
3) нарушения коронарного кровообращения;
4) расстройства функции перикарда.
Механизмы развития при сердечной недостаточности
При любой форме поражения сердца с момента его возникновения в организме развиваются компенсаторные реакции, направленные на предупреждение развития общей недостаточности кровообращения. Наряду с общими «внесердечными» механизмами компенсации при недостаточности сердца включаются компенсаторные реакции, осуществляющиеся в самом сердце. К ним относятся:
1) расширение полостей сердца с увеличением их объема (тоногенная дилятация) и увеличение ударного объема сердца;
2) учащение сердечных сокращений (тахикардия);
3) миогенная дилятация полостей сердца и гипертрофия миокарда.
Нарушения энергетического обмена в миокарде могут быть результатом недостаточности окисления, развития гипоксии, уменьшения активности ферментов, участвующих в окислении субстратов, и разобщения окисления и фосфорилирования.
Недостаточность субстратов для окисления чаще всего возникает вследствие уменьшения кровоснабжения сердца и изменения состава притекающей к сердцу крови, а также нарушения проницаемости клеточных мембран.
Склероз коронарных сосудов является наиболее частой причиной уменьшения кровоснабжения сердечной мышцы. Относительная ишемия сердца может быть результатом гипертрофии, при которой увеличение объема мышечных волокон не сопровождается соответствующим увеличением числа кровеносных капилляров.
Метаболизм миокарда может быть нарушен как при недостатке (например, гипогликемия), так и при избытке (например, при резком увеличении в притекающей крови молочной, пировиноградной кислот, кетоновых тел) некоторых субстратов. Вследствие сдвига рН миокарда возникают вторичные изменения активности ферментных систем, приводящие к нарушениям метаболизма.
На молекулярном и клеточном уровне механизмы патогенеза СН едины при самых разных причинах и формах СН: Нарушение энергообеспечения миокарда:
Повреждение мембран и ферментных систем кардиомиоцитов→ Дисбаланс ионов и жидкости в кардиомиоцитах→ Расстройства нейро-гуморальной регуляции сердца→ Снижение силы и скорости сокращений и расслаблений миокарда→ Развитие СН
Алкогольная полинейропатия выявляется у 20-30% больных алкоголизмом. Больные предъявляют жалобы на; парестезии в дистальных отделах конечностей, чаще ног (ощущения “ползания мурашек”, ‘онемения”, “стягивания”, “покалывания” и т.п.). Иногда отмечаются разнообразные болевые ощущения вплоть до острых стреляющих болей, напоминающих таковые при спинной сухотке, или крайне неприятные для больного ощущения жжения в подошвах, усиливающиеся при афферентной стимуляции последних. При объективном исследовании выявляется полиневритический тип расстройств обычно всех видов чувствительности, иногда сочетающийся с явлениями гиперпатии.
Развивается мышечная слабость, начиная с дистальных групп мышц на ногах. В связи с этим, а также из-за нарушения глубокой чувствительности затрудняется походка, появляется ощущение “ватности ног”. Иногда развивается крампи — тонические болезненные спазмы отдельных групп мышц. Сухожильные рефлексы, особенно на ногах, снижаются. Ахилловы рефлексы не вызываются в 80-80%, а коленные — в 50% случаев алкогольной полинейропатии. Следует отметить, что значительное снижение ахилловых рефлексов может наблюдаться у больных алкоголизмом без других явных признаков полинейропатии. Мышечный тонус в паретичных группах мышц снижен, а позже развивается и атрофия последних. Характерны также вазодистонические и нейротрофические изменения в виде мраморности кожных покровов, гипергидроза, отечности мягких тканей на голенях, стопах и кистях. При электронейромиографии выявляются признаки поражения периферического мотонейрона и ухудшение проводимости по нервным волокнам.
Наряду с полинейропатией у больных алкоголизмом развиваются также мононевриты, в патогенезе которых помимо факторов токсико-метаболического характера, связанных с употреблением алкоголя, играют роль и другие воздействия: травматизация, переохлаждение и т.п. Типично развитие травматических мононевритов, наиболее часто – лучевого нерва, в результате сдавления последнего массой тела во время сна в состоянии алкогольного опьянения.
Результаты патоморфологических исследований свидетельствуют о наличии двух основных механизмов повреждения нервных волокон при алкоголизме: аксональной дегенерации и демиелинизации. Аксональная дегенерация связана с токсическим воздействием метаболита этанола — ацетальдегида, а также пировиноградной кислоты. В патогенезе демиелинизации основное значение имеет дефицит витаминов группы В и никотиновой кислоты, В связи с этим при лечении алкогольных поражений периферической нервной системы широко применяется парентеральное введение больших доз витаминов группы В и никотиновой кислоты.
Поражение мышц при алкоголизме определяется как алкогольная миопатия. Она имеет острую и хроническую формы. Острая алкогольная миопатия развивается на фоне тяжелого запоя и проявляется остро возникающей слабостью в мышцах проксимальных отделов конечностей, сопровождающейся болезненностью, тоническими спазмами, отеками ног и миоглобинурией. Хронический вариант алкогольной миопатии проявляется нарастающей слабостью в проксимальных отделах мышц конечностей, чаще в нижних, диффузными или локальными мышечными спазмами, выраженной болезненностью мышц.
В патогенезе заболевания важная роль принадлежит токсическому воздействию этанола на клеточную мембрану и митохондрии мышечной ткани, при этом отмечаются выраженные нарушения окислительно-восстановительных процессов, белкового синтеза и обмена кальция в мышцах. При алкогольной миопатии в крови отмечается повышение уровней креатинфосфокиназы и альдолазы, причем наибольшее повышение выявляется при остром проявлении заболевания. Появляющаяся миоглобинурия в ряде случаев приводит к развитию острой почечной недостаточности из-за поражения почек.
При патоморфологическом исследовании мышц при острой алкогольной миопатии выявляется картина острого мышечного некроза с деструкцией мышечных волокон и внутриклеточным отеком. При хроническом течении заболевания изменения в мышцах менее выражены и представлены деструктивными процессами в ряде мышечных волокон с признаками регенерации. При ЭМГ исследовании с использованием локального метода отведения выявляется уменьшение амплитуды и длительности потенциалов действия двигательных единиц и рост числа полифазных потенциалов в измененных мышцах.
У лиц, злоупотребляющих спиртными напитками, на фоне острой алкогольной интоксикации в сочетании с голоданием выявляются субклинические признаки алкогольной миопатии в виде повышения уровня креатинфосфокиназы в крови и незначительных изменений при ЭМГ исследовании и биопсии мышц.
У больных алкоголизмом некротические изменения мышц могут развиться также в результате их сдавления массой тела в состоянии алкогольного опьянения, что определяется как синдром позиционного сдавления. По клиническим проявлениям и биохимическим изменениям данный синдром близок к острой алкогольной миопатии, но помимо признаков некроза мышц, как правило, имеются также повреждения кожных покровов и нейропатии травматического, и компрессионно-ишемического характера.
Лечение алкогольных миопатий предусматривает прекращение употребления спиртных напитков и проведение, дезинтоксикационной терапии (глюкозо-новокаиновая смесь, гемодез, реополиглюкин, раствор соды, трентал, мочегонные препараты). При острой алкогольной миопатии и синдроме позиционного сдавления следует проводить мероприятия, направленные на предупреждение, развития острой почечной недостаточности.
Источник
Сердце — это орган с высокими энергетическими потребностями и гибкостью метаболизма, что позволяет использовать множество энергетических субстратов для производства АТФ в различных физиологических условиях. Zhang, Fernandez-Caggiano и McCommis с соавторами сходятся во мнении, что митохондриальный переносчик пирувата является ключевым метаболическим узлом для поддержания обмена веществ в миокарде и важнейшей детерминантой гибкости метаболизма миокарда при сердечной недостаточности.
Основным источником энергии для выработки АТФ в сердечной мышце является окисление жирных кислот. Вклад глюкозы как субстрата, а также гликолиза в базовый метаболизм миокарда составляет около 30 %. Другие альтернативные субстраты, в том числе аминокислоты, лактат и кетоновые тела, вносят лишь скромный вклад в производство базального АТФ в здоровом сердце у взрослых. В здоровом миокарде взрослого человека вклад аминокислот, молочной кислоты и кетоновых тел как альтернативных источников окисления и получения АТФ очень скромный.
Усиленный метаболизм глюкозы в клетке в отсутствие кислорода впервые описал в 1861 году Луи Пастер на примере дрожжей. Позднее была обнаружена связь этого процесса с анаэробным гликолизом, что назвали «эффектом Пастера» [1]. Впоследствии Отто Варбург обнаружил, что опухолевые клетки вырабатывают огромное количество молочной кислоты путем анаэробного гликолиза. Это явление, суть которого ученый верно описал как характеристику опухолевого метаболизма, было названо «эффектом Варбурга» (исследователь неверно полагал, что открытый им феномен является главной причиной и движущей силой процесса образования опухолей) [2]. Недавние исследования связывают существование эффекта Варбурга в опухолевых клетках с возросшей потребностью в синтезе биомассы и поддержании жизнеспособности клеток. Это открывает новые перспективы использования модуляторов метаболизма в качестве методов лечения опухолей [3, 4].
Первичной причиной изменения метаболизма миокарда в течение эпизодов ишемии является нехватка кислорода, что выражается в нарушении процессов окисления и функций митохондрий и приводит к ускорению утилизации глюкозы кардиомиоцитами путем анаэробного гликолиза, накоплению пировиноградной кислоты и превращению последней в лактат. Однако аналогичные эффекты вследствие изменения экспрессии генов и активности ферментов были обнаружены при сердечной недостаточности даже при отсутствии гипоперфузии и ишемии. Среди прочих факторов эти изменения связывают с повышением «метаболического» стресса стенки миокарда [5], активацией программы экспрессии фетальных генов [6] и накоплением липотоксичных продуктов обмена жирных кислот [7, 8]. Учитывая более низкую эффективность производства АТФ на моль глюкозы при анаэробном гликолизе, чем при аэробном окислении, остается загадкой, почему метаболизм миокарда идет по пути анаэробного гликолиза, приводящего в итоге к меньшему количеству энергии, в условиях, когда достаточно и кислорода, и субстрата.
Функциональный митохондриальный переносчик пирувата (МПП), состоящий из двух субъединиц (MPC1 и MPC2), крайне важен для внутриклеточного гомеостаза, так как он обеспечивает транспорт пирувата внутрь митохондрии. Тем самым поддерживается функционирование цикла трикарбоновых кислот (ЦТК) путем введения в него субстратов в виде ацетил-кофермента А [9]. Впервые биохимическая функция MПП была описана в 1971 году [10], после чего появились сообщения о снижении активности МПП в раковых клетках с сопутствующим уменьшением окисления пирувата [11]. Утрата функции МПП в неопластических клетках способствует росту опухоли, свидетельствуя о том, что нарушенная активность МПП может по крайней мере частично объяснять эффект Варбурга и его влияние на метаболизм раковых клеток [12]. Кроме того, захват пирувата митохондриями с помощью МПП необходим для эффективного глюконеогенеза в печени и регуляции уровня глюкозы в крови [13].
В статье из «Nature Metabolism» приведены результаты работы трех независимых групп исследований. Все ученые сходятся во мнении, что нарушение регуляции работы МПП в миокарде при сердечной недостаточности является ключевым пунктом в разделении путей гликолиза и аэробного окисления глюкозы [14–16]. Изучая миокард при сердечной недостаточности, Fernandez-Caggiano с соавт. [15] наблюдали снижение уровня MPC1 в клетках сердечной мышцы — эффект, который был получен в животных моделях гипертрофии сердца, — и остановку сердца после инфузии ангиотензина II или поперечного сужения аорты (ПСА). Специфичная для сердца делеция в генах, кодирующих MPC1 или MPC2, приводит к гипертрофии миокарда, сердечной недостаточности и преждевременной смерти, а также к нарушению утилизации пирувата в митохондриях миокарда. Как результат чрезмерной экспрессии MPC1, стимулирующей тем самым формирование и активность MPC в сердце, Fernandez-Caggiano и др. наблюдали улучшение сердечной функции и снижение гипертрофии после ПСА, что указывает на то, что пируватный транспорт в митохондрии является критическим центром для поддержания метаболизма и функций миокарда во время сердечных приступов и нарушения адаптации обмена веществ в миокарде.
На мышиной модели с генетической делецией гена MPC1 (и его отсутствием в миокарде) Zhang с соавт. [14] независимо от других групп исследователей наблюдали развитие гипертрофии миокарда, сердечной недостаточности и преждевременной смерти. Они провели фенотипирование путем метаболомики образцов сердечной мышцы (патологически измененной от сердечной недостаточности) с дефицитом MPC1. Исследователи выявили признаки чрезмерной зависимости миокарда от глюкозы (признак доминирования гликолиза) и сниженного биотока углерода в митохондрии для включения его в состав метаболитов ЦТК. Также было обнаружено накопление метаболитов анаболизма, таких как лактат, пируват, аминокислоты и промежуточные продукты пентозо-фосфатного пути, наряду с повышенным синтезом гликогена. Стремясь перестроить метаболизм миокарда, минуя процессы гликолиза и переноса пирувата внутрь митохондрий, Zhang с соавт. содержали мышей с сердечной недостаточностью в условиях кетогенной диеты или диеты с высоким содержанием жиров. У этих мышей сердечная недостаточность была следствием отсутствия MPC1. Итог применения этих диет был в остановке ремоделирования миокарда. Эти преимущества, однако, не являются панацеей, потому что после эпизода повреждения миокарда применение кетогенной диеты не предотвращает наступление исходов ПСА. Только когда кетогенная диета используется в течение трех недель до ПСА, можно наблюдать протективный эффект для миокарда.
Независимо от описанных выше данных, McCommis с соавт. [16] наблюдали сходные перестройку метаболизма миокарда и исходы у мышей с кардиоспецифичным отсутствием MPC2 (делецией кардиоспецифичного гена MPC2). Также функция сердца при сердечной недостаточности при отсутствии MPC2 восстанавливалась с помощью кетогенной диеты. При оценке фракций ацилкарнитина путем метаболомики на разных группах мышей, которых кормили нормально или содержали в условиях кетогенной диеты, выявили критическое накопление средне- и длинноцепочечных ацилкарнитинов у мышей, употреблявших обычный корм. Проводя оценку диет с различным содержанием жиров, McCommis с соавт. обнаружили, что последствия недостаточности МПП в миокарде можно предотвратить или даже остановить с помощью диеты с высоким содержанием жира (диеты с низким содержанием жира и среднецепочечных триацилглицеридов не давали такого эффекта). Помимо кетогенной диеты, краткосрочное голодание с кетозом и (в меньшей степени) непосредственное использование как пищевой добавки кетоновых тел (инъекции β-гидроксибутирата или добавка в виде кетонового эфира) также препятствовали ремоделированию миокарда. Подробные транскрипционные, протеомные и метаболомные анализы выявили, что причина этих эффектов не в первичном метаболизме кетоновых тел, а в усиленном окислении жирных кислот.
В совокупности эти исследования показывают, что центральная роль в регулировании базального обмена веществ в миокарде и его функции принадлежит утилизации пирувата митохондриями. Также выделяют потенциальную роль диеты, способствующей повышению метаболизма жирных кислот, которую можно применять с целью остановки ремоделирования миокарда и восстановления его функции.
Кетоновые тела (β-гидроксибутират, ацетоацетат, ацетон) синтезируются в митохондриях печени из ацетил-кофермента А, полученного, в свою очередь, из жирных кислот. В условиях, когда углеводы ограничены, например, при голодании или низкоуглеводной диете, кетоновые тела служат альтернативным источником энергии. В тканях, где преобладает окислительное фосфорилирование (окисление субстратов в присутствии кислорода), таких как сердце, мозг и мышцы, происходит окисление кетоновых тел с образованием ацетил-кофермента А в качестве субстрата для ЦТК [17, 18].
Невзирая на то, что окисление кетоновых тел лишь незначительно способствует выработке энергии в условиях базального обмена веществ, несколько недавних исследований показали, что метаболизм кетоновых тел существенно влияет на развитие сердечной недостаточности. В соответствии с выводами описываемых исследований, обнаружилось, что диета с высоким содержанием жиров тормозит ремоделирование миокарда. Такие результаты были получены в ходе исследований на животных моделях гипертонической болезни и повреждения сердца, вызванного перегрузкой давлением [19, 20]. После перегрузки сердца давлением у животных с нарушенным процессом окисления кетоновых тел были выявлены признаки прогрессирования процесса патологического ремоделирования миокарда и ухудшения сердечной деятельности [21, 22]. В противовес этому, у животных с усиленным метаболизмом кетоновых тел (как следствия повышенной экспрессии генов окисления кетоновых тел) наблюдалась лучшая переносимость повреждения миокарда вследствие перегрузки давлением [23]. В совокупности эти исследования подтверждают кардиопротективную роль кетоновых тел в условиях стресса миокарда и служат основой для внесения изменений в диету, предложенных Fernandez-Caggiano, Zhang, McCommis и др.
Усиленный процесс гликолиза в условиях недостатка аэробного окисления глюкозы не может компенсировать сниженное энергоснабжение в миокарде после ишемического повреждения сердечной мышцы и в условиях сердечной недостаточности [24]. Подавление МПП, важнейшего регулятора потока пирувата от гликолиза (в цитоплазме) в митохондрии, может служить объяснением преобладания анаэробного гликолиза в миокарде даже в условиях нормоксии (эффект Варбурга при сердечной недостаточности). Липолиз в периферических тканях увеличивается при сердечной недостаточности вследствие хронической катехоламиновой стимуляции. Это, в свою очередь, приводит к циркуляции повышенной концентрации свободных жирных кислот [7, 25]. Нарушение окисления жирных кислот при сердечной недостаточности происходит вследствие уменьшения захвата карнитина белком-переносчиком (карнитин-ацилкарнитин транслоказой — митохондриальным белком, который осуществляет перенос ацилированного карнитина в матрикс митохондрии). Это приводит к повышению уровней циркулирующих ацилкарнитинов и снижению β-окисления жирных кислот.
Повышение доступности и утилизации кетоновых тел при сердечной недостаточности и поддержание метаболизма кетоновых тел на оптимальном уровне, похоже, является защитным механизмом в гипертрофированном миокарде и при сердечной недостаточности [21–23, 26], поэтому кетоновые тела могут служить в качестве альтернативного источника энергии с кардиопротективным эффектом. Тем не менее, даже несмотря на то, что кетоновые тела можно с легкостью окислить, а их высокая концентрация повышает скорость работы ЦТК и концентрацию его метаболитов, пищевые добавки с кетоновыми телами недостаточны для повышения нарушенной эффективности энергетического метаболизма при сердечной недостаточности [27, 28]. Согласно данным этих трех исследований, хроническая кетогенная внутренняя среда, а не резкое увеличение содержания кетонов в крови, по-видимому, оказывает благоприятное воздействие на миокард при сердечной недостаточности благодаря повышению окисления жирных кислот. Но даже в таком случае сердечная деятельность так и остается неполноценной. Это нарушение метаболизма миокарда можно объяснить ухудшением аэробного окисления глюкозы вследствие снижения уровня МПП, что ограничивает ток пирувата в митохондрии. Исследования влияния кетогенной терапии на течение сердечной недостаточности у людей в настоящее время ограничены, однако интересен факт увеличения концентрации промежуточных продуктов метаболизма кетоновых тел у пациентов с сахарным диабетом, которых лечат натрий-зависимыми ингибиторами котранспортера глюкозы 2-го типа.
Несмотря на то, что результаты этих трех исследований позволили лучше понять влияние МПП на метаболизм миокарда при сердечной недостаточности, ряд вопросов еще остается без ответов. Неясно, как контролируются уровни и функции МПП в здоровом миокарде, а как — при сердечной недостаточности: путем транскрипционной, трансляционной или посттрансляционной регуляции. Кроме того, еще предстоит выяснить, как влияет комплекс МПП на активацию программы экспрессии фетальных генов.
С биохимической точки зрения неизвестно, как специфика метаболизма кетоновых тел усиливает окисление жирных кислот. Предполагается, что из кетоновых тел образуются промежуточные метаболиты ЦТК (например, оксалоацетат), что может привести к повышению его скорости. Сопутствует этому повышенный захват жирных кислот митохондриями за счет активации фермента карнитин-пальмитоилтрансферазы I. В таком случае определенные уровни и регуляторные функции малонил-кофермента А и цитрата могут связывать два процесса (ЦТК и биосинтез жирных кислот).
Вышеописанные результаты свидетельствуют, что определенные кетогенные диеты могут служить в качестве поддерживающего немедикаментозного способа лечения людей с сердечной недостаточностью. Можно предполагать, что дефицит энергии в миокарде при сердечной недостаточности можно модифицировать, используя субстратную специфичность и терапевтическую модуляцию обмена веществ. Лучшее понимание взаимосвязи между метаболизмом кетоновых тел и жирных кислот в условиях хронического кетогенеза может помочь определить конкретные цели для непосредственного усиления окисления жирных кислот и устойчивого обеспечения энергией сердца при сердечной недостаточности (Рис. 1).
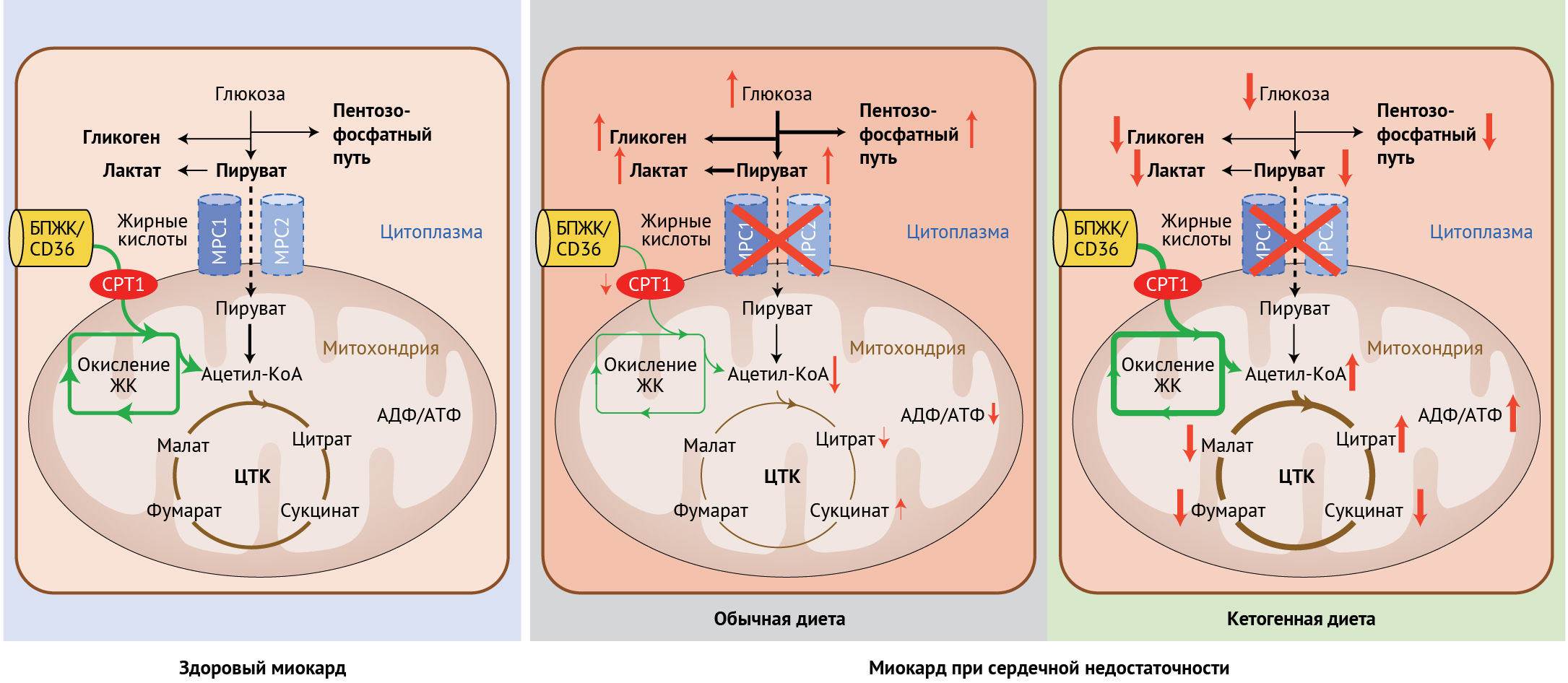
Рисунок 1 | Обмен промежуточных метаболитов в здоровом миокарде и при сердечной недостаточности, а также роль кетогенной диеты в коррекции субстратного метаболизма при сердечной недостаточности
Обмен веществ в миокарде в первую очередь зависит от окисления жирных кислот (примерно 70 % от всего произведенного АТФ). Вклад глюкозы как субстрата, а также гликолиза в базовый метаболизм миокарда составляет около 30 % (рисунок слева). При сердечной недостаточности окисление жирных кислот нарушено, а усиленный процесс гликолиза не может компенсировать сниженный уровень аэробного окисления глюкозы. Подавление митохондриального переносчика пирувата (МПП) снижает ток пирувата из гликолиза (в цитоплазме) в митохондрии даже в условиях нормоксии. Таким образом, происходит накопление лактата, пирувата и промежуточных продуктов пентозо-фосфатного пути, а также сам ЦТК производит меньшее количество АТФ (средний рисунок). Кетогенная диета ускоряет окисление жирных кислот и протекание ЦТК, приводя, таким образом, к увеличению синтеза АТФ. Концентрации промежуточных метаболитов гликолиза, лактата и гликогена (рисунок справа) сохраняются приближенными к норме.
Сокращения: FATP/CD36 — белок-переносчик жирной кислоты; CPT1 — фермент карнитин-пальмитоилтрансфераза 1.
Нашли опечатку? Выделите фрагмент и нажмите Ctrl+Enter.
Источник