Раас в патогенезе сердечной недостаточности

При ослаблении сократительной функции сердечной мышцы и появлении застойных явлений в организме больного норадреналин, высвобождающийся из окончаний симпатических нервов, возбуждая b1-адренорецепторы клеток юкстагломерулярного аппарата, стимулирует секрецию ренина. Другим стимулом секреции ренина является снижение почечного кровотока в результате вызванной катехоламинами (через а-адренорецепторы) констрикции эфферентных артериол почечных клубочков.
Позже усугубить эти механизмы может диуретическая терапия, которая способствует увеличению транспорта натрия хлорида через стенку почечных канальцев.
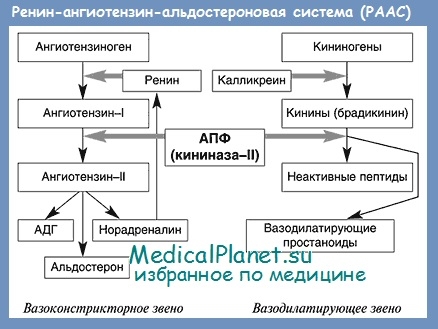
Известно, что ренин-ангиотензин-альдостероновая система (РААС) играет важную роль в регуляции артериального давления и водно-электролитного обмена. В последние годы получены убедительные доказательства того, что чрезмерная активация РААС, которая обнаруживается у большинства больных ХСН, является независимым от других факторов показателем неблагоприятного прогноза.
Ангиотензин II играет центральную роль в РААС. В частности, он является мощным вазоконстриктором, несколькими путями повышает артериальное давление, вызывает тахикардию, приводит к ремоделированию миокарда. Ангиотензин II способствует задержке в организме натрия и воды. Эти действия прямо или косвенно опосредуются через симпатическую нервную систему, антидиуретический гормон, альдостерон или угнетение активности блуждающего нерва.
Ангиотензин II оказывает существенное влияние на миокардиоциты и участвует в ремоделировании миокарда как после инфаркта, так и при других заболеваниях. Ремоделирование миокарда, развивающееся в этих случаях, является непременным фактором, определяющим течение и исходы ХСН.
Роль ангиотензина II в развитии ХСН существенно повышается при наличии у больных атеросклеротических изменений в коронарных артериях. В таких случаях ангиотензин II способствует усиленному окислению ЛПНП, ускоренному прогрессированию атеросклероза, появлению нестабильных атеросклеротических бляшек. Известно, что ангиотензин II образуется непосредственно в тканях, в том числе в сердце и сосудах, где, как и во многих других органах, имеется собственная ангиотензиновая система.
В пораженных атеросклерозом артериях эта система особенно активна (Н.А. Грацианский).
Ангиотензин II способствует также образованию супероксида кислорода, который приводит к разрушению важнейшего вазодилатирующего фактора – NО (окиси азота). Вследствие этого снижается вазодилатирующая функция сосудистого эндотелия, возникают ишемические изменения в миокарде, снижается его сократительная функция.
Совокупность этих изменений может быть причиной разрушения или разрыва атеросклеротических бляшек, возникновения вазоспастических явлений в сердце, что может вызывать обострение ИБС, появление нестабильных форм стенокардии и инфаркта миокарда, внезапную коронарную смерть, развитие острой и хронической сердечной недостаточности.
Ангиотензин II считается главным эффекторным пептидом РААС. Почти все известные эффекты активации РААС в крови, органах и тканях обусловлены влиянием ангиотензина П на его специфические рецепторы.
Большое практическое значение имеет то, что образование главного эффекторного пептида РААС – ангиотензина II – может происходить без участия как ангиотензин I-превращающего фермента, так и ренина.
Подавляя активность АПФ с помощью ингибитора АПФ, можно снизить уровень ангиотензина II и тем самым повлиять на механизмы обратной связи. Благодаря этому ингибиторы АПФ способны оказывать положительное влияние на течение сердечной недостаточности.
Компенсаторный характер нейрогуморальных изменений, повышение активности симпатической и ангиотензин-альдостероновой систем на раннем этапе застойной сердечной недостаточности заключаются в том, что возникают вазоконстрикция, компенсаторная тахикардия, гипертрофия миокарда и умеренная (по старой терминологии – тоногенная) дилатация сердца.
Адаптивный характер этих изменений проявляется, в частности, в том, что активизация РААС при начинающейся сердечно-сосудистой недостаточности способствует поддержанию артериального давления на уровне, обеспечивающем достаточный кровоток и перфузию кислорода в жизненно важных органах. Однако по мере прогрессирования заболевания компенсаторные реакции постепенно дают противоположные результаты. Длительная активация РААС приводит к прогрессированию заболевания и ухудшению прогноза.
Гиперактивация симпатического отдела нервной системы сопровождается повышением в плазме крови уровня норадреналина, что также обусловливает вазоконстрикцию, усиление тахикардии, задержку в организме натрия и воды. Нарастание симпатической активности может в определенной мере ослабляться вследствие того, что активность адренергических рецепторов саморегулируется по механизму отрицательной обратной связи. При этом, однако, может нарушаться функция барорецепторов, что вызывает дальнейшее повышение активности САС.
Повышенный уровень норадреналина в плазме крови является следствием увеличения его секреции с одновременным снижением клиренса. Норадреналин плазмы крови экскретируется в основном почками. При сердечной недостаточности в результате снижения клубочковой фильтрации клиренс норадреналина снижается и соответственно увеличивается его уровень в плазме. Ингибиторы АПФ могут снижать содержание норадреналина в плазме крови и нормализовать опосредованные САС рефлексы. Возможно, это происходит благодаря саморегуляции активности адренергических рецепторов.
Многими исследователями показана бесспорная связь между высоким уровнем циркулирующего норадреналина и повышенной летальностью больных с хронической застойной сердечной недостаточностью. Отрицательное воздействие избытка катехоламинов на сердце обусловлено многими факторами: прямым кардиотоксическим эффектом, непрямым влиянием за счет активации нейрогуморальных механизмов и повышения общего периферического сопротивления, активизацией свертывающей системы крови, нарушением функции бета-адренорецепторов, частыми нарушениями сердечного ритма и т.д.
Чрезмерная симпатическая активность вызывает повреждение миокардиоцитов вплоть до их некроза.
При сердечной недостаточности возрастает активность барорецепторов, что ведет к повышению симпатической стимуляции сердца и периферических сосудов. Ангиотензин II может вызывать еще большее повышение симпатической активности.
Определенное значение в патогенезе сердечной недостаточности имеет изменение концентрации в крови предсердного натрийуретического фактора. Известно, что этот фактор секретируется миокардом предсердий и желудочков. Повышение напряжения стенок предсердий или желудочков, обычно связанное с ростом давления наполнения соответствующих камер сердца, является основным стимулом для увеличенной секреции предсердного натрийуретического фактора.
Предсердный натрийуретический фактор способствует дилатации артерий, он снижает вазоконстрикторный эффект, обусловленный другими нейрогормонами, стимулирует экскрецию натрия и воды из организма. При сердечной недостаточности выраженность этого вазодилататорного эффекта снижается из-за вазоконстрикторных эффектов других гормонов и потенциально положительное влияние предсердного натрийуретического фактора на функцию почек ослабевает.
Еще одно из звеньев патогенеза сердечной недостаточности связано с изменением содержания в крови антидиуретического гормона. Повышенная активность симпатико-адреналовой и ренин-ангиотензин-альдостероновой систем стимулирует секрецию антидиуретического гормона клетками задней доли гипофиза. Повышение уровня ангиотензина II также может стать важным стимулом увеличения секреции антидиуретического гормона.
Антидиуретический гормон резко уменьшает экскрецию воды почками, увеличивает ее реабсорбцию в дистальных канальцах и собирательных трубочках, значительно усиливает задержку воды в организме. Кроме того, он может вызвать распространенную вазоконстрикцию. На фоне высокой концентрации в крови больных с застойной сердечной недостаточностью антидиуретического гормона резко снижается экскреция натрия и выводы почками, нарастают отеки, увеличивается жажда (часто становится нестерпимой). Очень высокое содержание антидиуретического гормона часто выявляется у больных с сердечной недостаточностью, которые длительно лечатся большими дозами диуретиков-салуретиков.
Следует учитывать, что с помощью ингибиторов АПФ можно существенно снизить уровень антидиуретического гормона.
– Вернуться в раздел нашего сайта “кардиология”
Оглавление темы “Болезни сердца и перикарда”:
- Эксудативный перикардит – клиника, диагностика
- Адгезивный перикардит – клиника, диагностика
- Бактериальные и вирусные перикардиты – клиника, диагностика
- Ревматические, уремические, посттравматические перикардиты – клиника, диагностика
- Гидроперикард и перикардиальные кисты – клиника, диагностика
- Опухоли сердца – клиника, диагностика
- Лечение перикардита – пункция перикарда
- Сердечная недостаточность – определение, эпидемиология
- Причины и механизмы развития сердечной недостаточности
- Ренин-ангиотензин-альдостероновая система (РААС) при сердечной недостаточности
Источник
Согласно современным представлениям, одно из основных значений в патогенезе развитии ХСН отводится ренин-ангиотензин-альдостероновая системе, все функции которой осуществляются через ангиотензин II. На рубеже 90-х годов с помощью типирования, клонирования обнаружено, что действие ангиотензина П возможно лишь при воздействии на ангиотензиновые рецепторы.
Ангиотензин II – важный компонент ренин-ангиотензин-альдостероновой системы
Ангиотензин II является наиболее важным эффекторным пептидом ренин-ангиотензин-альдостероновой системы вызывает вазоконстрикцию, способствует симпатической трансмиссии, а также задержке солей и воды в почках. Также показана роль ангиотензина II в развитии центральной нервной системы, дифференциации и регенерации. Ангиотензин II действует на структуры мозга по обе стороны гемато-энцефалического барьера, что определяет его способность вызывать жажду, модулировать симпатическую активность на периферии, ослаблять барорецепторный рефлекс, вызывать натрийурез и высвобождать вазопрессин и другие гипофизарные гормоны. Эффекты ангиотензина II при сердечной недостаточности включают гипертрофию левого желудочка сердца и структурные изменения сосудистой сети, сердца и почек (например, формирование неоинтимы, постинфарктное ремоделирование, нефросклероз.
Рецепторы к ангиотензину (AT1 и АТ2) – структурные компоненты ренин-ангиотензин-альдостероновой системы
Идентифицированы два основных подтипа рецепторов к ангиотензину II: тип 1 (AT1) и тип 2 (АТ2). Оба подтипа имеют много общего, так как обладают аналогичной геометрией, при которой они содержат полипептидные цепи с около 360 аминокислотами, формирующими семикратно сложенную трансмембранную гирлянду, соединенную при активации с внутриклеточными G-протеинами. Но здесь гомогенность заканчивается, так как определяется гомология только 30 % аминокислотной последовательности. Более того, AT1-рецепторы генетически связаны с хромосомой 3, в то время как ген рецептора к ангиотензину II локализован в Х-хромосоме. В организме животных описаны также подтипы 3 и 4 ангиотензиновых рецепторов, однако, у человека эти рецепторы не были обнаружены.
Несмотря на то, что рецепторы к ангиотензину II принадлежат к одному семейству рецепторов, их сигнальные механизмы и функции различаются.
Рецепторы AT1 фактически ответственны за все известные действия ангиотензина II.
Ренин-ангиотензин-альдостероновая система – Эффекты ангиотензина II, реализуемые через AT1-рецепторы (по R. Willenheimer, 1999):
- Вазоконстрикция
- Задержка жидкости и натрия (продукция альдостерона)
- Гипертрофия миоцитов и гладкомышечных клеток
- Фиброз миокардиальной и сосудистой стенки (синтез и отложение
коллагена) - Гиперплазия фибробластов
- Цитотоксические влияния на миокард
- Увеличение секреции эндотелина-1
- Повышение высвобождения вазопрессина
- Активация симпатической нервной системы
- Стимулирование образования супероксида
- Увеличение уровня ингибитора активатора плазминогена
- Изменение генной экспрессии
Их роль была хорошо документирована после выделения рецептора и определения локализации гена. Связывание ангиотензина II с AT1-рецептором приводит к последующей реализации большинства периферических и центральных эффектов ангиотензина II на артериальное давление, осморегуляцию и клеточный рост, и, таким образом, опосредует патофизиологическое действие ренин-ангиотензин-альдостероновой системы при заболеваниях сердца, сосудов и почек. Распределение рецепторов AT1 у взрослых людей повсеместное, включая сосудистую сеть, почки, надпочечники, сердце, печень и головной мозг. Они вовлечены в механизмы вазоконстрикции, сократимости миокарда, высвобождения альдостерона и вазопрессина, задержки солей в почках, механизмы сосудистой и сердечной гипертрофии.
О сигнальных механизмах и фармакологических эффектах рецепторов АТ2 известно значительно меньше. Если в фетальных тканях они присутствуют повсеместно, то у взрослых в высоких концентрациях определяются только в надпочечниках, матке, яичниках, сосудистом эндотелии и некоторых участках головного мозга. Повышенная экспрессия или доминирование АТ2-рецепторов происходит при патофизиологических состояниях, таких как хроническая сердечная недостаточность, после перенесенного инфаркта миокарда, заболеваниях почек и нервной системы. Предполагается, что АТ2-рецепторы вовлечены в ингибирование пролиферации клеток и, возможно, в клеточную дифференцировку, развитие, регенерацию и даже программированную клеточную смерть (апоптоз).
Проведенные исследования подтверждают гипотезу, что АТ2-рецепторы опосредуют антипролиферацию при изучении поврежденных сонных артерий крысы. Обнаружено, что формирование неоинтимы снижалось на 70 % после трансфера гена АТ2 – рецептора. Аналогично, перенос гена АТ2-рецепторов в культуру гладкомышечных клеток аорты крысы уменьшал пролиферацию клеток. Таким образом, вышеприведенные данные предполагают, что стимулирование АТ2-рецепторов может ингибировать и, возможно, вызывать обратное развитие способствующих росту эффектов ангиотензина II через AT1-рецепторы. Кроме того, ангиотензин II воздействием на АТ2-рецепторы способствует развитию нейронов в различных клеточных линиях нейронного происхождения. Параллельно с этими морфологическими изменениями, стимулирование АТ2-рецепторов модулирует экспрессию важных белковых волокон и, таким образом, прямо вовлечено в развитие нейронов путем реорганизации цитоскелета.
Предполагается, что ангиотензин II может взаимодействовать с системой брадикинин/NO по АТ2-рецепторзависимому механизму, улучшая эндотелиальную и миокардиальную функции. В проведенных экспериментальных работах ангиотензин II стимулировал продукцию цГМФ в интерстициальной жидкости почек крысы и в аорте. Эти эффекты предупреждаются ингибированием синтазы NO, а также блокадой рецепторов к брадикинину Б2.
Экспериментальные исследования показывают, что пептид ангиотен-зин(1-7) образуется как из ангиотензина I,так и из ангиотензина II под влиянием эндопептидазы. Этот пептид вызывает повышение содержания NO путем стимуляции специфических AT1-7 рецепторов или АТ2-рецепторов. Более того, через AT1-рецепторы ангиотензин II вызывает формирование супероксида, который разрушает оксид азота. Следовательно, во время блокады AT1-рецепторов увеличивается содержание оксида азота вследствие снижения образования оксида азота.
Таким образом, повышенная активность NO, наблюдаемая при лечении ингибиторами ангиотензин-превращающего фермента, также присутствует во время терапии с антагонистами AT1-рецепторов. Основываясь на экспериментальных данных, антагонисты AT1-рецепторов обладают потенциалом для более эффективного противодействия неблагоприятной нейрогуморальной активации при ХСН, чем ИАПФ.
Точная оценка влияния ИАПФ на компоненты ренин-ангиотензин-альдостероновой системы всегда представляла трудности, так как АПФ является в основном тканевым ферментом, более чем 90 % которого образуется и действует в эндотелии легочных сосудов. Поэтому было показано, что эффекты ИАПФ не всегда параллельны способности этих препаратов подавлять активность ангиотензинпревращающего фермента, определяемую в плазме крови, т.е. эффекты всех ИАПФ имеют как бы два уровня: плазменный, проявляющийся немедленно после начала терапии, и тканевой – действующий через недели и даже месяцы после начала лечения.
Образование ангиотензина II происходит разными путями и под влиянием различных нейрогормонов. Как стало ясно, только 10-15 % ангиотензина II синтезируется при непосредственном участии АПФ и лишь эта часть образующегося ангиотензина II может контролироваться ИАПФ. Значительно большая часть ангиотензина II синтезируется под влиянием других ферментов – химазы, тонина, катепсина G, пептидилдипептидазы эндотелиальных клеток, почечной карбоксипептидазы. Кроме того, образование ангиотензина II может происходить и непосредственно из ангиотензиногена, минуя образование ангиотензина I как промежуточного продукта. Из этого следует, что влияние ИАПФ на образование ангиотензина II является неспецифическим и неполным.
В связи с этим можно предположить, что и синтез альдостерона не может полностью контролироваться применением ИАПФ. Поэтому большой интерес вызывает применение ингибиторов активных компонентов ренин-ангиотензин-альдостероновой системы – антагонистов рецепторов ангиотензина II и конкурентных антагонистов альдостерона.
Таким образом, ренин-ангиотензин-альдостероновая система играет важнейшую роль в патогенезе ХСН (хронической сердечной недостаточности).
Диуретики влияют на систему РААС.
Полезно знать- Современные принципы терапии собак, кошек и других мелких животных с застойной сердечной недостаточностью (ХСН)
- Современные подходы к применению диуретиков (мочегонных препаратов) в терапии хронической сердечной недостаточности
- Нейрогуморальная концепция патогенеза хронической сердечно-сосудистой недостаточности
Источник
В основе сердечной недостаточности могут лежать различные этиологические и патогенетические факторы. Одни вызывают непосредственное повреждение миокарда, другие – увеличение нагрузки на сердце.
Согласно современной нейрогуморальной концепции патогенеза сердечной недостаточности, основная роль в ее развитии отводится симпатоадреналовой и ренин-ангиотензин-альдостероновой системам и противостоящей им системе предсердного натрийуретического фактора. Нейрогуморальная система вовлекается в патологический процесс уже на ранних этапах сердечной недостаточности. Ее активация, с одной стороны, способствует компенсации сердечной деятельности в ответ на снижение сердечного выброса, а с другой – стимулирует прогрессирование декомпенсации необратимых изменений в организме. Активация нейрогуморальной системы является важнейшим патогенетическим фактором СН, маркером наличия и тяжести заболевания, мишенью для терапевтических воздействий. При развитии СН происходят существенные сдвиги баланса между вазоконстрикторами, вазодилататорами и факторами роста с преобладанием вазоконстрикторов. Среди них особого внимания заслуживают эндотелины – вазоактивные пептиды, состоящие из 21 аминокислоты.
Известно 3 изоформы эндотелина, однако наиболее выраженными вазоконстриктивными свойствами обладает эндотелин-1. Эндотелин-1 повышает содержание Са2+ в гладкомышечных клетках сосудов (вазоконстрикция), оказывает митогенный и пролиферативный эффект, стимулирует секрецию и увеличение активности ряда ключевых гормонов: альдостерона, АДГ, ангиотензина-2. Вазоконстрикторный эффект ЭТ-1 приводит к повышению периферического сосудистого сопротивления, а также сопротивлению сосудов сердца, мозга и почек. Биологические эффекты эндотелинов реализуются через 2 типа эндотелиновых рецепторов: рецепторы типа А обнаруживается преимущественно в гладкомышечных клетках сосудов и вовлечены в передачу вазоконстрикторных и митогенных эффектов ЭТ-1 и рецепторы типа В, локализующиеся в эндотелии и участвующие в вазодилатации благодаря вызываемому ими высвобождению простациклина и оксида азота и высвобождении альдостерона из коры надпочечников.
У больных сердечнососудистыми заболеваниями уровень ЭТ-1 постоянно увеличен и, возможно, нарушены функции эндотелиновых рецепторов. У пациентов с острым коронарным синдромом и инфарктом миокарда уровень ЭТ-1 всегда повышен и возвращается к нормативным значениям через 72 часа. Вместе с тем, при наличии таких осложнений как кардиогенный шок, сердечная недостаточность и дисфункция левого желудочка уровень ЭТ-1 остается повышенным и служит маркером тяжести состояния и предиктором неблагоприятного прогноза в отдаленные сроки и низкой выживаемости больных.
Дисфункция миокарда (снижение насосной функции сердца) вначале бессимптомная. Для предотвращения ее прогрессирования с развертыванием полной клинической картины сердечной недостаточности запускаются различные компенсаторные механизмы, однако со временем мощность их оказывается недостаточной. Более того, со временем они начинают приобретать отрицательное значение, приводя к прогрессированию сердечной недостаточности. Включение компенсаторных механизмов и постепенная трансформация их роли из положительной в отрицательную роль и есть основа патогенеза хронической сердечной недостаточности (ХСН).
Компенсаторные механизмы при ХСН принято делить на кардиальные (сердечные) и экстракардиальные (внесердечные)
Различают следующие кардиальные компенсаторные механизмы:
1. Адренергическая стимуляция силы сердечных сокращений (инотропные резервы, резервы сократимости). Она направлена на увеличение УО и, следовательно, – на увеличение МОС
2. Адренергическая стимуляция ЧСС (хронотропные резервы), направленная на поддержание МОС.
3. Гиперфункция миокарда, несвязанная с адренергической стимуляцией. В соответствии с двумя типами сокращения миофибрилл различают и два типа компенсаторной гиперфункции: изометрическую и изотоническую.
Изометрическая гиперфункция характеризуется увеличением напряжения без изменения длины мышечного волокна, сопровождается значительным усилением коронарного кровотока и потребления кислорода. Амплитуда сердечных сокращений и МОС существенно не меняются. Работа сердца возрастает за счет увеличения систолического давления, дилатация полости сердца отсутствует; выражена гипертрофия миокарда. Компенсация достигается за счет повышения сократимости миокарда. Изометрическая гиперфункция встречается при перегрузке сопротивлением (гипертоническая болезнь, стеноз аортального клапана и др.).
Изотоническая гиперфункция (базируется на механизме Франка-Старлинга) характеризуется уменьшением длины мышечного волокна без изменения напряжения. Она проявляется незначительным увеличением коронарного кровотока и потребления кислорода. Амплитуда сокращений миокарда значительно увеличена. Работа сердца возрастает за счет увеличения МОС без существенного изменения артериального давления. Дилатация соответствующей полости значительно выражена. Компенсация достигается за счет увеличения конечного диастолического давления в желудочке. Этот тип гиперфункции встречается при перегрузке объемом: увеличение ОЦК, клапанная недостаточность
При равной внешней работе сердца изотоническая гиперфункция осуществляется с меньшей затратой энергии, чем изометрическая, поэтому энергетически более выгодна и обратима. Любая гиперфункция сердца приводит к увеличению интенсивности функционирования структур (ИФС): увеличивается потребление кислорода на единицу массы миокарда, возрастает интенсивность процессов окислительного фосфорилирования, больше образуется АТФ. Все это активирует генетический аппарат клетки с развитием компенсаторной гипертрофии.
4. Гипертрофия миокарда – увеличение массы миокардиальных клеток без увеличения их числа. Она направлена на приведение структуры в соответствие с возросшей функцией (ИФС равна 1). Компенсаторная гипертрофия миокарда протекает в 3 стадии:
– аварийная стадия. Активация генетического аппарата кардиомиоцитов вызывает усиление синтеза структурных и сократительных белков, что приводит к увеличению массы миокарда. Но ИФС еще не достигает нормы, так как массы миокарда недостаточно (ИФС>1);
– стадия завершившейся гипертрофии и относительно устойчивой гиперфункции. В эту стадию наблюдается полное соответствие массы миокарда и возросшей функции, т.е. повышенная функция равномерно распределяется по увеличенной массе миокарда, и ИФС вновь равна 1;
– стадия прогрессирующего кардиосклероза. Она характеризуется замещением кардиомиоцитов соединительной тканью. Причина – нарушение энергетического обмена в кардиомиоцитах и активация гликолиза, что приводит к накоплению недоокисленных продуктов цикла Кребса и к ацидозу. Соединительная ткань не может выполнять сократительную функцию, поэтому наступает декомпенсация сердечной деятельности (ИФС<1).
Перечисленные кардиальные компенсаторные механизмы тесно связаны между собой, а также регулируются местными приспособительными реакциями. Они заключаются в выделении клетками миокарда и коронарных сосудов медиаторов. Одни из них оказывают положительный инотропный и вазоконстрикторный эффект (катехоламины, ангиотензин-2, эндотелин, серотонин, тромбоксан А2), другие – отрицательный инотропный и вазодилатирующий эффект (релаксирующий фактор эндотелия коронаров, предсердный натрийуретический пептид, простагландины I2 и Е2).
Экстракардиальные компенсаторные механизмы СН включают активацию симпатоадреналовой системы (САС) и ренин-ангиотензин-альдостероновой системы (РААС).
1. Активация симпатоадреналовой системы. Механизмы активации САС при сердечной недостаточности, вероятно, связаны с гипоперфузией тканей и повышением венозного давления (патологические барорецепторные рефлексы). Активация САС приводит к реализации сосудосуживающих эффектов катехоламинов (адреналина и норадреналина), что необходимо для поддержания АД и перераспределения кровотока в пользу жизненно важных органов (сердце, головной мозг), т.е. развития централизации кровообращения. Это возможно потому, что коронарные и церебральные сосуды не суживаются под влиянием катехоламинов. Кроме того, под действием катехоламинов происходит учащение и усиление сердечных сокращений (но эти компенсаторные эффекты относятся к сердечным).
2. Активация РААС осуществляется двумя путями. Во-первых, снижение АД, уменьшение МОС и рефлекторное сужение почечных артериол приводит к гипоперфузии почек. Происходит возбуждение волюморецепторов клеток ЮГА и усиливается выработка ренина. Во-вторых, высокие концентрации КХА в крови вызывают возбуждение β-адренорецепторов клеток ЮГА → усиление выработки ренина. Далее запускается вся система РААС: ренин → ангиотензин-1 → ангиотензин-2 → альдостерон → усиление реабсорбции натрия → гипернатриемия → повышение осмотического давления → возбуждение осморецепторов гипоталамуса → усиление выработки и высвобождения через нейрогипофиз вазопрессина → усиление реабсорбции воды. Цель активации РААС – поддержать МОС за счет увеличения ОЦК и вазоконстрикторного эффекта ангиотензина-2.
Однако компенсаторные механизмы, первоначально направленные на предотвращение развития сердечной недостаточности, в конечном итоге приводят к ее прогрессированию. Это происходит по тому, что на первый план начинают выступать неадекватность этих механизмов и их неблагоприятные эффекты. Фактически это ведет к декомпенсации сердечной деятельности.
К механизмам декомпенсации при сердечной недостаточности относятся:
1. Неадекватность тахикардии (которая исходно служит для увеличения МОС) заключается в укорочении диастолы и ухудшении вследствие этого коронарной перфузии и увеличении потребности миокарда в кислороде.
2. Неадекватность задержки натрия и жидкости (исходно направленной на поддержание МОС за счет увеличения ОЦК) приводит к развитию отеков и увеличению объемной нагрузки на сердце.
3. Неадекватности вазоконстрикции (исходно направленной на поддержание АД и сохранение перфузии жизненно важных органов) приводит к увеличению нагрузки на сердце вследствие роста общего сопротивления периферических сосудов и ухудшению кровоснабжения органов и тканей, особенно почек.
4. Неадекватность гипертрофии миокарда (изначально направленной на субстратное обеспечение возросшей функции) неизбежно ведет к декомпенсации сердечной деятельности. Почему? Гипертрофия миокарда при хронической сердечной недостаточности с самого начала рассматривается как несбалансированная форма роста на органном, тканевом, клеточном, субклеточном и молекулярном уровне.
– на уровне органа – масса сердца увеличивается значительно быстрее, чем образуются новые нервные окончания, в результате снижается плотность симпатической иннервации и содержание норадреналина, ухудшается адренергическая регуляция сердца. Первоначальная трата норадреналина для мобилизации сократительной функции сердца преобладает над его синтезом, вследствие чего содержание норадреналина в сердечной мышце уменьшается, и это становится причиной снижения инотропного действия симпатической нервной системы на сердце;
– на уровне ткани – происходит отставание роста количества капилляров от массы кардиомиоцитов, вследствие чего уменьшается примерно в 3 раза количество коронарных капилляров на единицу поперечного сечения миокарда, т.е. ухудшается питание (кровоснабжение) сердечной мышцы, а в итоге – ухудшается доставка кислорода и субстратов к гипертрофированному миокарду;
– на уровне клетки – снижение мощности мембранных ионообменных насосов, мембранных ферментных систем. Площадь сарколеммы увеличивается пропорционально квадрату, а объем клетки – пропорционально кубу, т.е. происходит относительное уменьшение площади мембраны, что играет ведущую роль в нарушении сокращения и расслабления сердечной мышцы;
– на уровне внутриклеточных органелл. Темпы увеличения массы митохондрий отстают от темпов увеличения массы миофибрилл, что закономерно ухудшает энергообеспечение кардиомиоцитов.
– на уровне молекулярных структур – снижается способность миозиновых волокон использовать энергию АТФ для сокращения, поскольку нарушается структура миозина и снижается его АТФазная активность.
5. Неадекватная нейрогуморальная регуляция, (изначально направленная на увеличение силы и частоты сердечных сокращений, что увеличивает МОС и повышает АД). Однако со временем приводит к следующим неблагоприятным последствиям:
▪ увеличение потребления кислорода сердцем под влиянием катехоламинов;
▪ увеличение нагрузки на сердце из-за вазоконстрикции и задержки в организме жидкости под влиянием катехоламинов, ангиотензина-2, альдостерона и вазопрессина;
▪ развитие отеков (из-за задержки воды и активации системы РААС);
▪ непосредственное неблагоприятное воздействие нейрогормонов на миокард:
– катехоламины увеличивают риск развития аритмий, вызывают перегрузку кардиомиоцитов кальцием (через активацию аденилатциклазы и увеличение внутриклеточной концентрации ЦАФ) и как следствие – формирование контрактур и некроза, стимулирование развития участков генерации; активируют факторы роста и синтез цитокинов и апоптоз;
– ангиотензин-2 и альдостерон суживают коронарные сосуды, ухудшая коронарный кровоток, стимулируют синтез коллагена и образование фиброзной ткани в миокарде, стимулируют ремоделирование.
Ремоделирование– неотъемлемая часть патогенеза ХСН. Под ремоделированием понимают процесс комплексного нарушения структуры и функции сердца в ответ на перегрузку или утрату части жизнеспособного миокарда. Процесс ремоделирования при ХСН включает в себя:
– прогрессирующее увеличение массы миокарда;
– дилатацию полостей сердца;
– изменение геометрических характеристик желудочков.
Зависимость функции сердца от его геометрии установил еще Гарвей в 17 веке. Однако этот термин стал использоваться в 80-х годах ХХ столетия. В норме в систолу сердце принимает эллипсоидную форму, а в диастолу – более сферическую. Подобное изменение геометрии во время сердечного цикла обеспечивает нормальную систолическую и диастолическую функцию. Нарушение этих геометрических характеристик при ремоделировании приводит к снижению фракции выброса, нарушениям системной гемодинамики и появлению клинических симптомов сердечной недостаточности (предшествует им и сопровождает их).
Вначале ремоделирование при сердечной недостаточности носит компенсаторный характер, а со временем ведет к ряду негативных последствий и, в конце концов, к срыву компенсации. Сердце вступает в фазу «прогрессирующего кардиосклероза и изнашивания структур» (по Ф.З. Меерсону), которая характеризуется следующими проявлениями: гибелью клеток, нарушением обновления структур, развитием склероза органа, прогрессирующим нарушением систолической и диастолической функции, биоэнергетики и увеличением риска развития аритмий.
Гибернация(hibernating – зимняя спячка) – состояние дисфункции левого желудочка в покое, вызванное его длительной гипоперфузией, частично или полностью исчезающее после улучшения коронарного кровообращения и/или снижения потребности миокарда в кислороде. При гибернации дисфункция левого желудочка более продолжительна, чем при острой ишемии миокарда и потенциально обратима, чем принципиально отличается от таковой у больных с постинфарктным кардиосклерозом.
Гибернирующий миокард представляет собой приспособительную реакцию, сущность которой заключается в том, что функция сердечной мышцы снижается до такой степени, что достигается равновесие между потребностями миокарда в кислороде и доставкой его кровью посредством коллатерального резидуального кровотока. Потребление кислорода миокардом снижается в ответ на уменьшение коронарного кровотока.
Благодаря тому, что на новом уровне поддерживается равновесие между потребностями миокарда в кислороде и доставкой его с кровью, отсутствуют симптомы и признаки ишемии миокарда, поэтому не развивается острый инфаркт миокарда. В биохимическом отношении гибернирубщий миокард – это «гипометаболическое состояние для сохранения энергии». При морфологическом исследовании зон гибернирующего миокарда не удается выявить некротические изменения.
При гибернирующем миокарде кардиомиоциты остаются жизнеспособными в течение длительного вр